-
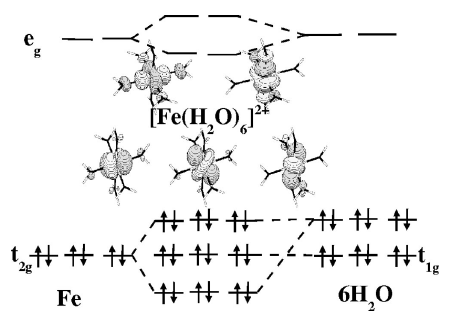
Comparison of density functionals for energy and structural differences between the high- [5T2g:(t2g)4(eg)2] and low- [1A1g:(t2g)6(eg)0] spin states of iron(II) coordination compounds. II. More functionals and the hexaminoferrous cation, [Fe(NH3)6]2+
A. Fouqueau, M.E. Casida, L.M. Lawson Daku, A. Hauser and F. Neese
Journal of Chemical Physics, 122 (4) (2005), p44110


DOI:10.1063/1.1839854 | unige:3272 | Abstract | Article HTML | Article PDF
The ability of different density functionals to describe the structural and energy differences between the high- [5T2g:(t2g)4(eg)2] and low- [1A1g:(t2g)6(eg)0] spin states of small octahedral ferrous compounds is studied. This work is an extension of our previous study of the hexaquoferrous cation, [Fe(H2O)6]2+, [J. Chem. Phys. 120, 9473 (2004)] to include a second compoundŌĆönamely, the hexaminoferrous cation, [Fe(NH3)6]2+ŌĆöand several additional functionals. In particular, the present study includes the highly parametrized generalized gradient approximations (GGAs) known as HCTH and the meta-GGA VSXC [which together we refer to as highly parametrized density functionals (HPDFs)], now readily available in the GAUSSIAN03 program, as well as the hybrid functional PBE0. Since there are very few experimental results for these molecules with which to compare, comparison is made with best estimates obtained from second-order perturbation theory-corrected complete active space self-consistent field (CASPT2) calculations, with spectroscopy oriented configuration interaction (SORCI) calculations, and with ligand field theory (LFT) estimations. While CASPT2 and SORCI are among the most reliable ab initio methods available for this type of problem, LFT embodies many decades of empirical experience. These three methods are found to give coherent results and provide best estimates of the adiabatic low-spinŌĆōhigh-spin energy difference, ╬öELHadia, of 12ŌĆŖ000ŌĆō13ŌĆŖ000ŌĆēcmŌłÆ1 for [Fe(H2O)6]2+ and 9ŌĆŖ000ŌĆō11ŌĆŖ000ŌĆēcmŌłÆ1 for [Fe(NH3)6]2+. All functionals beyond the purely local approximation produce reasonably good geometries, so long as adequate basis sets are used. In contrast, the energy splitting, ╬öELHadia, is much more sensitive to the choice of functional. The local density approximation severely over stabilizes the low-spin state with respect to the high-spin state. This ŌĆ£density functional theory (DFT) spin pairing-energy problemŌĆØ persists, but is reduced, for traditional GGAs. In contrast the hybrid functional B3LYP underestimates ╬öELHadia by a few thousands of wave numbers. The RPBE GGA of Hammer, Hansen, and N├Ėrskov gives good results for ╬öELHadia as do the HPDFs, especially the VSXC functional. Surprisingly the HCTH functionals actually over correct the DFT spin pairing-energy problem, destabilizing the low-spin state relative to the high-spin state. Best agreement is found for the hybrid functional PBE0.